Analysis Pipeline
RNAseq analysis pipeline¶
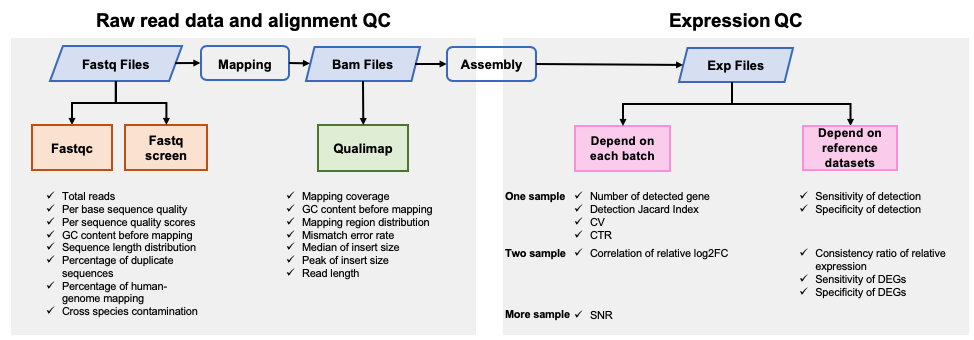
Preliminary processing of raw fastq reads was performed using fastp v0.19.6 to remove adapter sequences1. Read alignment and quantification was conducted using HISAT v2.1, SAMtools v1.3.1, StringTie v1.3.4 and Ballgown v2.14.12. Reference human genome build 38 and gene model from Ensembl were used for read mapping and gene quantification. log2 transformation was then conducted based on Fragments Per Kilobase of transcript per Million mapped reads (FPKM) values. To avoid infinite values, a value of 0.01 was added to the FPKM value of each gene before log2 transformation. Expression profiles based on detected genes were used for further analysis. A gene was considered detectable (expressed) in a biological group within a batch if ≥ 3 reads were mapped onto it in at least two of the three replicates.
Quality control analysis of sequencing data at pre-alignment and post-alignment level was conducted using FastQC v0.11.53, FastQ Screen v0.12.04, Qualimap v2.0.05, and MultiQC v1.86.
Results generated from this APP can be visualized by QDP report.
Reference¶
-
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. J Bioinformatics 34, i884-i890 (2018). ↩
-
Pertea, M., Kim, D., Pertea, G.M., Leek, J.T. & Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc 11, 1650 (2016). ↩
-
Andrews, S. FastQC: a quality control tool for high throughput sequence data https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. (2017). ↩
-
Wingett, S.W. & Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 7 (2018). ↩
-
García-Alcalde, F. et al. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 28, 2678-2679 (2012). ↩
-
Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047-3048 (2016). ↩