Analysis Pipeline for WGS & WES
Quality control pipeline for WGS¶
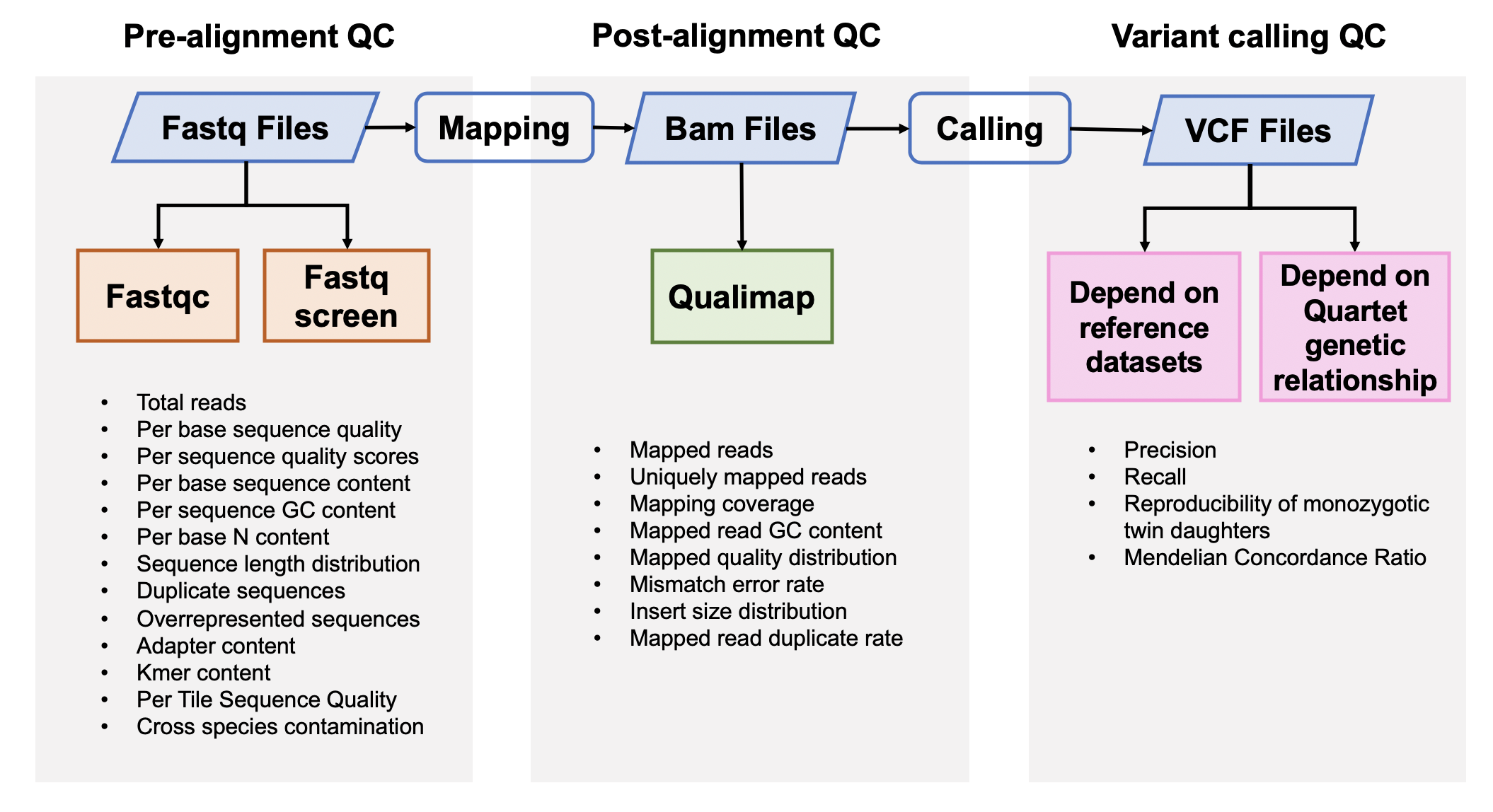
This Quartet quality control pipeline evaluate the performance of reads quality and variant calling quality. This pipeline accepts FASTQ format input files or VCF format input files. If the users input FASTQ files, this APP will output the results of pre-alignment quality control from FASTQ files, post-alignment quality control from BAM files and variant calling quality control from VCF files. GATK best practice pipelines (implemented by SENTIEON software) were used to map reads to the reference genome and call variants. If the users input VCF files, this APP will only output the results of variant calling quality control.
Quartet quality control analysis pipeline started from FASTQ files is implemented across seven main procedures:
- Pre-alignment QC of FASTQ files
- Genome alignment
- Post-alignment QC of BAM files
- Germline variant calling
- Variant calling QC depended on benchmark sets of VCF files
- Check Mendelian inheritance states across four Quartet samples of every variants
- Variant calling QC depended on Quartet genetic relationship of VCF files
Quartet quality control analysis pipeline started from VCF files is implemented across three main procedures:
- Variant calling QC depended on benchmark sets of VCF files
- Check Mendelian inheritance states across four Quartet samples of every variants
- Variant calling QC depended on Quartet genetic relationship of VCF files
Results generated from this APP can be visualized by QDP report.
1. Pre-alignment QC of FASTQ files¶
Fastqc v0.11.5¶
FastQC is used to investigate the quality of fastq files
Fastq Screen 0.12.0¶
Fastq Screen is used to inspect whether the library were contaminated. For example, we expected 99% reads aligned to human genome, 10% reads aligned to mouse genome, which is partly homologous to human genome. If too many reads are aligned to E.Coli or Yeast, libraries or cell lines are probably comtminated.
fastq_screen --aligner <aligner> --conf <config_file> --top <number_of_reads> --threads <threads> <fastq_file>
2. Genome alignment¶
sentieon-genomics:v2019.11.28¶
Reads were mapped to the human reference genome GRCh38 using Sentieon BWA.
${SENTIEON_INSTALL_DIR}/bin/bwa mem -M -R "@RG\tID:${group}\tSM:${sample}\tPL:${pl}" -t $nt -K 10000000 ${ref_dir}/${fasta} ${fastq_1} ${fastq_2} | ${SENTIEON_INSTALL_DIR}/bin/sentieon util sort -o ${sample}.sorted.bam -t $nt --sam2bam -i -
3. Post-alignment QC¶
Qualimap and Paicard Tools (implemented by Sentieon) are used to check the quality of BAM files. Deduplicated BAM files are used in this step.
Qualimap 2.0.0¶
qualimap bamqc -bam <bam_file> -outformat PDF:HTML -nt <threads> -outdir <output_directory> --java-mem-size=32G
Sentieon-genomics:v2019.11.28¶
${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${Dedup_bam} --algo CoverageMetrics --omit_base_output ${sample}_deduped_coverage_metrics --algo MeanQualityByCycle ${sample}_deduped_mq_metrics.txt --algo QualDistribution ${sample}_deduped_qd_metrics.txt --algo GCBias --summary ${sample}_deduped_gc_summary.txt ${sample}_deduped_gc_metrics.txt --algo AlignmentStat ${sample}_deduped_aln_metrics.txt --algo InsertSizeMetricAlgo ${sample}_deduped_is_metrics.txt --algo QualityYield ${sample}_deduped_QualityYield.txt --algo WgsMetricsAlgo ${sample}_deduped_WgsMetricsAlgo.txt
4. Germline variant calling¶
HaplotyperCaller implemented by Sentieon is used to identify germline variants.
${SENTIEON_INSTALL_DIR}/bin/sentieon driver -r ${ref_dir}/${fasta} -t $nt -i ${recaled_bam} --algo Haplotyper ${sample}_hc.vcf
5. Variants Calling QC¶
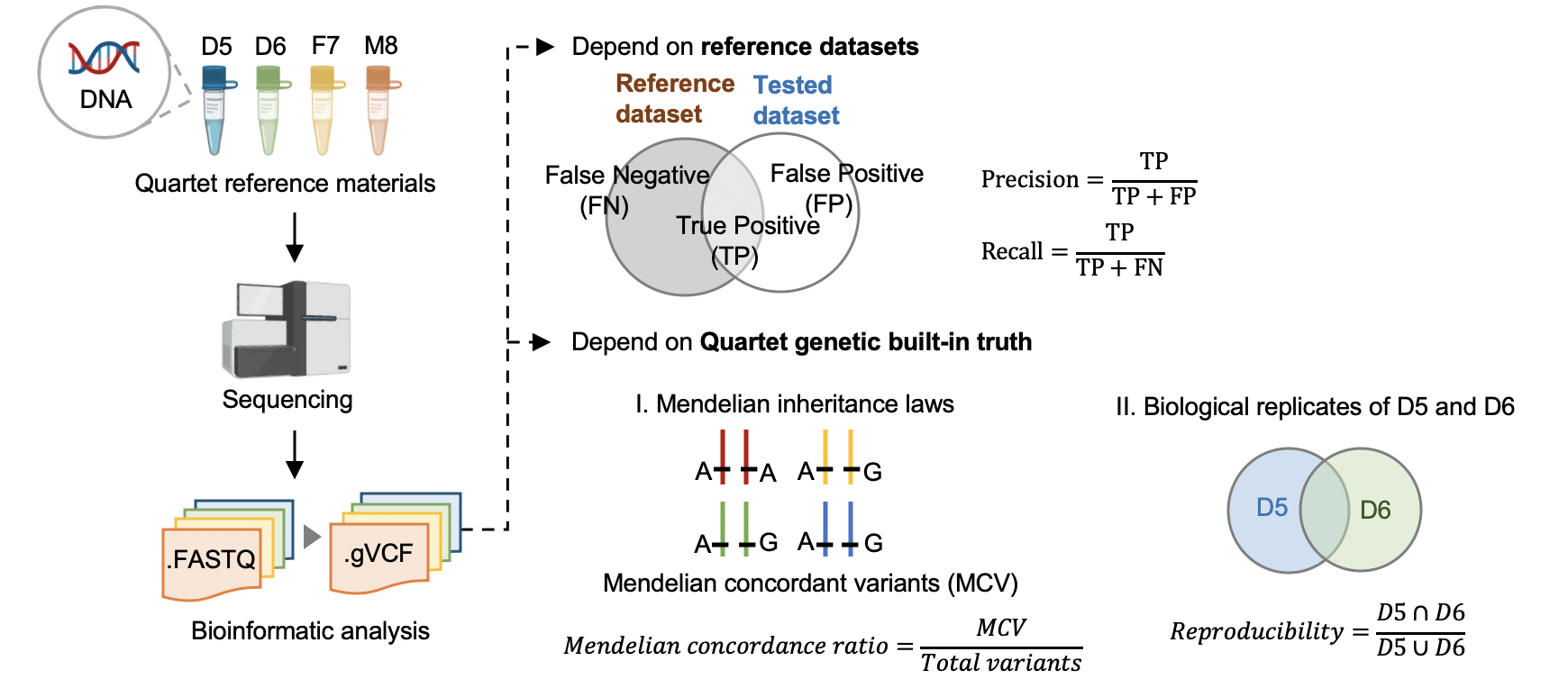
5.1 Performance assessment based on benchmark sets¶
Hap.py v0.3.9¶
Variants were compared with benchmark calls in benchmark regions.
5.2 Performance assessment based on Quartet genetic built-in truth¶
VBT v1.1¶
We splited the Quartet family to two trios (F7, M8, D5 and F7, M8, D6) and then do the Mendelian analysis. A Quartet Mendelian concordant variant is the same between the twins (D5 and D6) , and follow the Mendelian concordant between parents (F7 and M8). Mendelian concordance rate is the Mendelian concordance variant divided by total detected variants in a Quartet family. Only variants on chr1-22,X are included in this analysis.
vbt mendelian -ref <fasta_file> -mother <family_merged_vcf> -father <family_merged_vcf> -child <family_merged_vcf> -pedigree <ped_file> -outDir <output_directory> -out-prefix <output_directory_prefix> --output-violation-regions -thread-count <threads>
Input files¶
1. Start from Fastq files¶
sample_id,project,fastq_1_D5,fastq_2_D5,fastq_1_D6,fastq_2_D6,fastq_1_F7,fastq_2_F7,fastq_1_M8,fastq_2_M8
# sample_id in choppy system
# project name
# oss path of D5 fastq read1 file
# oss path of D5 fastq read2 file
# oss path of D6 fastq read1 file
# oss path of D6 fastq read2 file
# oss path of F7 fastq read1 file
# oss path of F7 fastq read2 file
# oss path of M8 fastq read1 file
# oss path of M8 fastq read2 file
2. Start from VCF files¶
sample_id,project,vcf_D5,vcf_D6,vcf_F7,vcf_M8
# sample_id in choppy system
# project name
# oss path of D5 VCF file
# oss path of D6 VCF file
# oss path of F7 VCF file
# oss path of M8 VCF file
Output Files¶
1. extract_tables.wdl/extract_tables_vcf.wdl¶
(FASTQ) Pre-alignment QC: pre_alignment.txt
(FASTQ) Post-alignment QC: post_alignment.txt
(FASTQ/VCF) Variants calling QC: variants.calling.qc.txt
2. quartet_mendelian.wdl¶
(FASTQ/VCF) Variants calling QC: mendelian.txt
Output files format¶
1. pre_alignment.txt¶
| Column name | Description |
|---|---|
| Sample | Sample name |
| %Dup | Percentage duplicate reads |
| %GC | Average GC percentage |
| Total Sequences (million) | Total sequences |
| %Human | Percentage of reads mapped to human genome |
| %EColi | Percentage of reads mapped to Ecoli |
| %Adapter | Percentage of reads mapped to adapter |
| %Vector | Percentage of reads mapped to vector |
| %rRNA | Percentage of reads mapped to rRNA |
| %Virus | Percentage of reads mapped to virus |
| %Yeast | Percentage of reads mapped to yeast |
| %Mitoch | Percentage of reads mapped to mitochondrion |
| %No hits | Percentage of reads not mapped to genomes mentioned above |
2. post_alignment.txt¶
| Column name | Description |
|---|---|
| Sample | Sample name |
| %Mapping | Percentage of mapped reads |
| %Mismatch Rate | Mapping error rate |
| Mendelian Insert Size | Median insert size(bp) |
| %Q20 | Percentage of bases >Q20 |
| %Q30 | Percentage of bases >Q30 |
| Mean Coverage | Mean deduped coverage |
| Median Coverage | Median deduped coverage |
| PCT_1X | Fraction of genome with at least 1x coverage |
| PCT_5X | Fraction of genome with at least 5x coverage |
| PCT_10X | Fraction of genome with at least 10x coverage |
| PCT_30X | Fraction of genome with at least 30x coverage |
3. variants.calling.qc.txt¶
| Column name | Description |
|---|---|
| Sample | Sample name |
| SNV number | Total SNV number (chr1-22,X) |
| INDEL number | Total INDEL number (chr1-22,X) |
| SNV query | SNV number in benchmark region |
| INDEL query | INDEL number in benchmark region |
| SNV TP | True positive SNV |
| INDEL TP | True positive INDEL |
| SNV FP | False positive SNV |
| INDEL FP | True positive INDEL |
| SNV FN | False negative SNV |
| INDEL FN | False negative INDEL |
| SNV precision | Precision of SNV calls when compared with benchmark calls in benchmark regions |
| INDEL precision | Precision of INDEL calls when compared with benchmark calls in benchmark regions |
| SNV recall | Recall of SNV calls when compared with benchmark calls in benchmark regions |
| INDEL recall | Recall of INDEL calls when compared with benchmark calls in benchmark regions |
| SNV F1 | F1 score of SNV calls when compared with benchmark calls in benchmark regions |
| INDEL F1 | F1 score of INDEL calls when compared with benchmark calls in benchmark regions |
4 {project}.summary.txt¶
| Column name | Description |
|---|---|
| Family | Family name defined by inputed project name |
| Reproducibility_D5_D6 | Percentage of variants were shared by the twins (D5 and D6) |
| Mendelian_Concordance_Quartet | Percentage of variants were Mendelian concordance |